This pipeline computes the correlation between cancer subtypes identified by different molecular patterns and selected clinical features.
Testing the association between subtypes identified by 2 different clustering approaches and 3 clinical features across 45 patients, no significant finding detected with P value < 0.05.
-
CNMF clustering analysis on sequencing-based miR expression data identified 3 subtypes that do not correlate to any clinical features.
-
Consensus hierarchical clustering analysis on sequencing-based miR expression data identified 3 subtypes that do not correlate to any clinical features.
Table 1. Get Full Table Overview of the association between subtypes identified by 2 different clustering approaches and 3 clinical features. Shown in the table are P values from statistical tests. Thresholded by P value < 0.05, no significant finding detected.
|
Clinical Features |
Statistical Tests |
MIRseq CNMF subtypes |
MIRseq cHierClus subtypes |
| Time to Death | logrank test | 0.755 | 0.504 |
| AGE | ANOVA | 0.199 | 0.465 |
| GENDER | Fisher's exact test | 0.419 | 0.374 |
Table S1. Get Full Table Description of clustering approach #1: 'MIRseq CNMF subtypes'
| Cluster Labels | 1 | 2 | 3 |
|---|---|---|---|
| Number of samples | 9 | 16 | 20 |
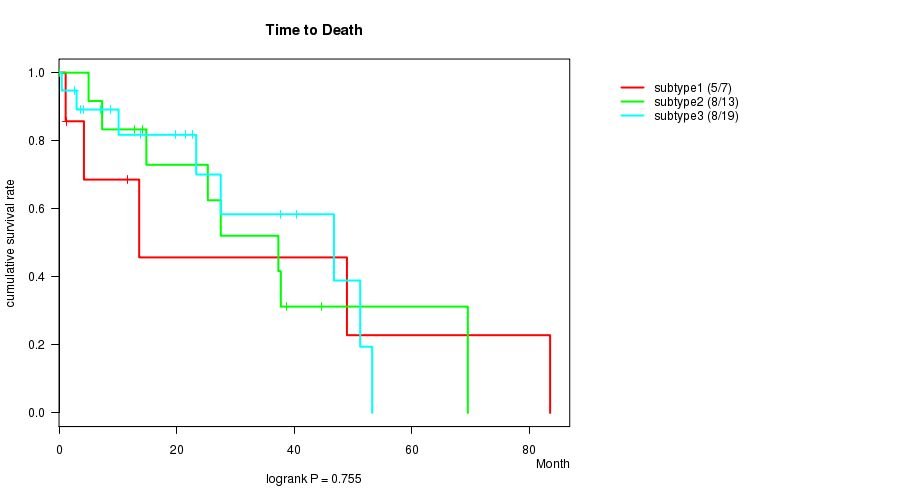
P value = 0.755 (logrank test)
Table S2. Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #1: 'Time to Death'
| nPatients | nDeath | Duration Range (Median), Month | |
|---|---|---|---|
| ALL | 39 | 21 | 0.1 - 83.6 (14.9) |
| subtype1 | 7 | 5 | 1.1 - 83.6 (11.6) |
| subtype2 | 13 | 8 | 0.1 - 69.6 (25.3) |
| subtype3 | 19 | 8 | 0.5 - 53.3 (19.8) |
Figure S1. Get High-res Image Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #1: 'Time to Death'
P value = 0.199 (ANOVA)
Table S3. Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #2: 'AGE'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 42 | 60.0 (15.9) |
| subtype1 | 7 | 54.9 (19.3) |
| subtype2 | 15 | 56.3 (17.0) |
| subtype3 | 20 | 64.7 (13.0) |
Figure S2. Get High-res Image Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #2: 'AGE'

P value = 0.419 (Fisher's exact test)
Table S4. Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #3: 'GENDER'
| nPatients | FEMALE | MALE |
|---|---|---|
| ALL | 18 | 27 |
| subtype1 | 2 | 7 |
| subtype2 | 8 | 8 |
| subtype3 | 8 | 12 |
Figure S3. Get High-res Image Clustering Approach #1: 'MIRseq CNMF subtypes' versus Clinical Feature #3: 'GENDER'

Table S5. Get Full Table Description of clustering approach #2: 'MIRseq cHierClus subtypes'
| Cluster Labels | 1 | 2 | 3 |
|---|---|---|---|
| Number of samples | 8 | 29 | 8 |
P value = 0.504 (logrank test)
Table S6. Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #1: 'Time to Death'
| nPatients | nDeath | Duration Range (Median), Month | |
|---|---|---|---|
| ALL | 39 | 21 | 0.1 - 83.6 (14.9) |
| subtype1 | 6 | 4 | 1.1 - 83.6 (12.6) |
| subtype2 | 26 | 14 | 0.1 - 69.6 (17.3) |
| subtype3 | 7 | 3 | 2.6 - 37.6 (21.4) |
Figure S4. Get High-res Image Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #1: 'Time to Death'

P value = 0.465 (ANOVA)
Table S7. Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #2: 'AGE'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 42 | 60.0 (15.9) |
| subtype1 | 6 | 58.2 (18.9) |
| subtype2 | 28 | 58.6 (15.9) |
| subtype3 | 8 | 66.4 (13.6) |
Figure S5. Get High-res Image Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #2: 'AGE'

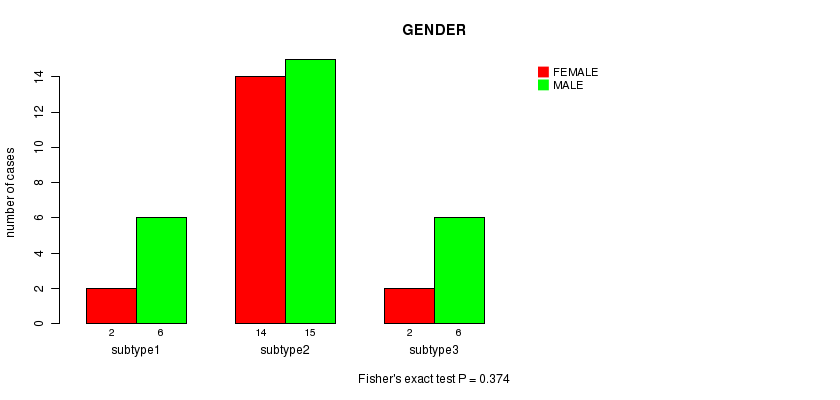
P value = 0.374 (Fisher's exact test)
Table S8. Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #3: 'GENDER'
| nPatients | FEMALE | MALE |
|---|---|---|
| ALL | 18 | 27 |
| subtype1 | 2 | 6 |
| subtype2 | 14 | 15 |
| subtype3 | 2 | 6 |
Figure S6. Get High-res Image Clustering Approach #2: 'MIRseq cHierClus subtypes' versus Clinical Feature #3: 'GENDER'
-
Cluster data file = LIHC.mergedcluster.txt
-
Clinical data file = LIHC.clin.merged.picked.txt
-
Number of patients = 45
-
Number of clustering approaches = 2
-
Number of selected clinical features = 3
-
Exclude small clusters that include fewer than K patients, K = 3
consensus non-negative matrix factorization clustering approach (Brunet et al. 2004)
Resampling-based clustering method (Monti et al. 2003)
For survival clinical features, the Kaplan-Meier survival curves of tumors with and without gene mutations were plotted and the statistical significance P values were estimated by logrank test (Bland and Altman 2004) using the 'survdiff' function in R
For continuous numerical clinical features, one-way analysis of variance (Howell 2002) was applied to compare the clinical values between tumor subtypes using 'anova' function in R
For binary clinical features, two-tailed Fisher's exact tests (Fisher 1922) were used to estimate the P values using the 'fisher.test' function in R
This is an experimental feature. The full results of the analysis summarized in this report can be downloaded from the TCGA Data Coordination Center.