(primary solid tumor cohort)
This pipeline computes the correlation between significantly recurrent gene mutations and selected clinical features.
Testing the association between mutation status of 7 genes and 8 clinical features across 99 patients, one significant finding detected with Q value < 0.25.
-
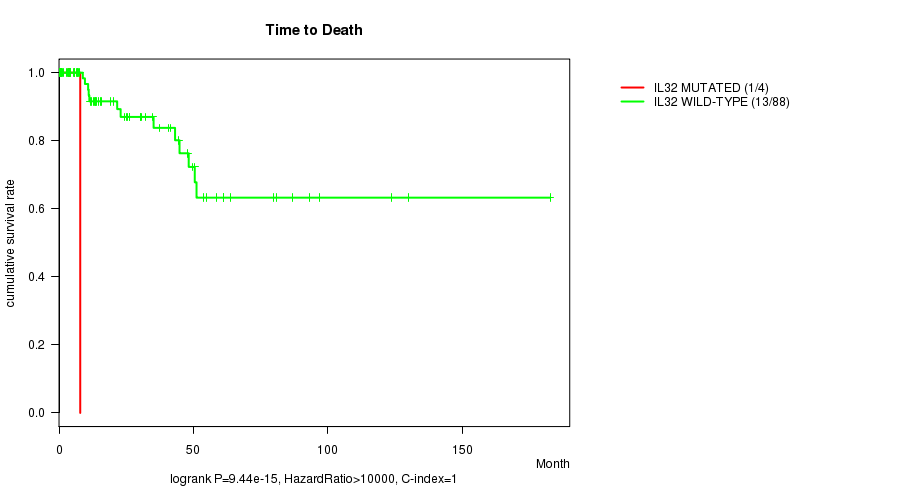
IL32 mutation correlated to 'Time to Death'.
Table 1. Get Full Table Overview of the association between mutation status of 7 genes and 8 clinical features. Shown in the table are P values (Q values). Thresholded by Q value < 0.25, one significant finding detected.
|
Clinical Features |
Time to Death |
AGE | GENDER |
KARNOFSKY PERFORMANCE SCORE |
PATHOLOGY T |
PATHOLOGY N |
PATHOLOGICSPREAD(M) |
TUMOR STAGE |
||
| nMutated (%) | nWild-Type | logrank test | t-test | Fisher's exact test | t-test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | |
| IL32 | 4 (4%) | 95 |
9.44e-15 (4.25e-13) |
0.37 (1.00) |
0.301 (1.00) |
0.783 (1.00) |
0.222 (1.00) |
0.671 (1.00) |
||
| CDC27 | 4 (4%) | 95 |
0.598 (1.00) |
0.782 (1.00) |
0.593 (1.00) |
0.783 (1.00) |
0.129 (1.00) |
0.452 (1.00) |
||
| MET | 7 (7%) | 92 |
0.695 (1.00) |
0.877 (1.00) |
1 (1.00) |
1 (1.00) |
1 (1.00) |
0.892 (1.00) |
||
| PCF11 | 7 (7%) | 92 |
0.834 (1.00) |
0.399 (1.00) |
0.423 (1.00) |
0.749 (1.00) |
0.202 (1.00) |
0.58 (1.00) |
1 (1.00) |
|
| SFRS2IP | 5 (5%) | 94 |
0.226 (1.00) |
0.121 (1.00) |
0.657 (1.00) |
0.107 (1.00) |
1 (1.00) |
0.0839 (1.00) |
0.328 (1.00) |
|
| NF2 | 6 (6%) | 93 |
0.598 (1.00) |
0.139 (1.00) |
1 (1.00) |
0.301 (1.00) |
0.0583 (1.00) |
0.0252 (1.00) |
0.0619 (1.00) |
|
| LGI4 | 4 (4%) | 95 |
0.19 (1.00) |
0.913 (1.00) |
0.301 (1.00) |
0.783 (1.00) |
0.707 (1.00) |
0.671 (1.00) |
P value = 9.44e-15 (logrank test), Q value = 4.2e-13
Table S1. Gene #3: 'IL32 MUTATION STATUS' versus Clinical Feature #1: 'Time to Death'
| nPatients | nDeath | Duration Range (Median), Month | |
|---|---|---|---|
| ALL | 92 | 14 | 0.0 - 182.7 (14.4) |
| IL32 MUTATED | 4 | 1 | 3.6 - 7.9 (4.8) |
| IL32 WILD-TYPE | 88 | 13 | 0.0 - 182.7 (15.1) |
Figure S1. Get High-res Image Gene #3: 'IL32 MUTATION STATUS' versus Clinical Feature #1: 'Time to Death'
-
Mutation data file = KIRP-TP.mutsig.cluster.txt
-
Clinical data file = KIRP-TP.clin.merged.picked.txt
-
Number of patients = 99
-
Number of significantly mutated genes = 7
-
Number of selected clinical features = 8
-
Exclude genes that fewer than K tumors have mutations, K = 3
For survival clinical features, the Kaplan-Meier survival curves of tumors with and without gene mutations were plotted and the statistical significance P values were estimated by logrank test (Bland and Altman 2004) using the 'survdiff' function in R
For continuous numerical clinical features, two-tailed Student's t test with unequal variance (Lehmann and Romano 2005) was applied to compare the clinical values between tumors with and without gene mutations using 't.test' function in R
For binary or multi-class clinical features (nominal or ordinal), two-tailed Fisher's exact tests (Fisher 1922) were used to estimate the P values using the 'fisher.test' function in R
For multiple hypothesis correction, Q value is the False Discovery Rate (FDR) analogue of the P value (Benjamini and Hochberg 1995), defined as the minimum FDR at which the test may be called significant. We used the 'Benjamini and Hochberg' method of 'p.adjust' function in R to convert P values into Q values.
This is an experimental feature. The full results of the analysis summarized in this report can be downloaded from the TCGA Data Coordination Center.