(primary solid tumor cohort)
This pipeline computes the correlation between significantly recurrent gene mutations and selected clinical features.
Testing the association between mutation status of 16 genes and 10 clinical features across 69 patients, 2 significant findings detected with Q value < 0.25.
-
SMAD2 mutation correlated to 'NUMBER.OF.LYMPH.NODES'.
-
ERBB2 mutation correlated to 'NUMBER.OF.LYMPH.NODES'.
Table 1. Get Full Table Overview of the association between mutation status of 16 genes and 10 clinical features. Shown in the table are P values (Q values). Thresholded by Q value < 0.25, 2 significant findings detected.
|
Clinical Features |
Time to Death |
AGE | GENDER |
HISTOLOGICAL TYPE |
PATHOLOGY T |
PATHOLOGY N |
PATHOLOGICSPREAD(M) |
TUMOR STAGE |
COMPLETENESS OF RESECTION |
NUMBER OF LYMPH NODES |
||
| nMutated (%) | nWild-Type | logrank test | t-test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | t-test | |
| SMAD2 | 5 (7%) | 64 |
0.497 (1.00) |
0.834 (1.00) |
1 (1.00) |
1 (1.00) |
0.525 (1.00) |
0.342 (1.00) |
1 (1.00) |
0.536 (1.00) |
1 (1.00) |
0.000868 (0.137) |
| ERBB2 | 4 (6%) | 65 |
0.273 (1.00) |
0.127 (1.00) |
1 (1.00) |
0.416 (1.00) |
0.453 (1.00) |
1 (1.00) |
0.662 (1.00) |
1 (1.00) |
0.000874 (0.137) |
|
| APC | 57 (83%) | 12 |
0.588 (1.00) |
0.897 (1.00) |
1 (1.00) |
0.634 (1.00) |
0.0753 (1.00) |
0.813 (1.00) |
0.362 (1.00) |
0.0491 (1.00) |
0.693 (1.00) |
0.495 (1.00) |
| KRAS | 38 (55%) | 31 |
0.0279 (1.00) |
0.534 (1.00) |
0.329 (1.00) |
0.27 (1.00) |
0.0973 (1.00) |
0.832 (1.00) |
0.327 (1.00) |
0.309 (1.00) |
0.0405 (1.00) |
0.34 (1.00) |
| TP53 | 45 (65%) | 24 |
0.924 (1.00) |
0.976 (1.00) |
0.803 (1.00) |
0.238 (1.00) |
0.0498 (1.00) |
0.935 (1.00) |
0.73 (1.00) |
0.297 (1.00) |
0.624 (1.00) |
0.476 (1.00) |
| SMAD4 | 8 (12%) | 61 |
0.447 (1.00) |
0.668 (1.00) |
1 (1.00) |
0.00581 (0.907) |
1 (1.00) |
0.645 (1.00) |
0.327 (1.00) |
0.594 (1.00) |
1 (1.00) |
0.586 (1.00) |
| KIAA1804 | 9 (13%) | 60 |
0.447 (1.00) |
0.24 (1.00) |
1 (1.00) |
1 (1.00) |
0.707 (1.00) |
1 (1.00) |
1 (1.00) |
0.957 (1.00) |
1 (1.00) |
0.0883 (1.00) |
| FBXW7 | 9 (13%) | 60 |
0.497 (1.00) |
0.92 (1.00) |
0.722 (1.00) |
1 (1.00) |
0.579 (1.00) |
0.199 (1.00) |
0.338 (1.00) |
0.341 (1.00) |
0.631 (1.00) |
0.721 (1.00) |
| NRAS | 5 (7%) | 64 |
0.116 (1.00) |
0.0221 (1.00) |
1 (1.00) |
1 (1.00) |
0.0521 (1.00) |
0.53 (1.00) |
0.555 (1.00) |
0.887 (1.00) |
1 (1.00) |
0.0288 (1.00) |
| TCF7L2 | 7 (10%) | 62 |
0.665 (1.00) |
0.744 (1.00) |
1 (1.00) |
1 (1.00) |
0.794 (1.00) |
0.85 (1.00) |
0.266 (1.00) |
0.514 (1.00) |
0.276 (1.00) |
0.21 (1.00) |
| PIK3CA | 7 (10%) | 62 |
0.497 (1.00) |
0.432 (1.00) |
1 (1.00) |
0.0348 (1.00) |
1 (1.00) |
0.62 (1.00) |
0.582 (1.00) |
0.63 (1.00) |
0.628 (1.00) |
0.0101 (1.00) |
| OPCML | 6 (9%) | 63 |
0.497 (1.00) |
0.966 (1.00) |
0.69 (1.00) |
1 (1.00) |
0.148 (1.00) |
1 (1.00) |
1 (1.00) |
0.504 (1.00) |
1 (1.00) |
0.724 (1.00) |
| SPATA8 | 3 (4%) | 66 |
0.194 (1.00) |
0.0775 (1.00) |
1 (1.00) |
1 (1.00) |
1 (1.00) |
1 (1.00) |
0.737 (1.00) |
1 (1.00) |
0.314 (1.00) |
|
| IL1RAPL2 | 5 (7%) | 64 |
0.765 (1.00) |
0.639 (1.00) |
1 (1.00) |
0.11 (1.00) |
1 (1.00) |
0.816 (1.00) |
1 (1.00) |
0.852 (1.00) |
1 (1.00) |
0.0153 (1.00) |
| FAM123B | 6 (9%) | 63 |
0.484 (1.00) |
0.485 (1.00) |
0.69 (1.00) |
1 (1.00) |
0.0463 (1.00) |
1 (1.00) |
0.207 (1.00) |
0.0851 (1.00) |
0.222 (1.00) |
0.639 (1.00) |
| ZIM3 | 5 (7%) | 64 |
0.069 (1.00) |
0.235 (1.00) |
1 (1.00) |
0.493 (1.00) |
0.366 (1.00) |
0.53 (1.00) |
1 (1.00) |
0.666 (1.00) |
0.0831 (1.00) |
0.00803 (1.00) |
P value = 0.000868 (t-test), Q value = 0.14
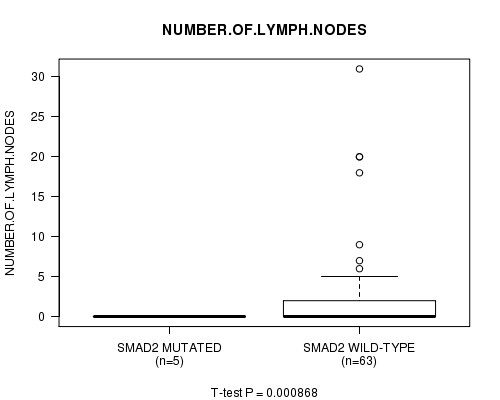
Table S1. Gene #11: 'SMAD2 MUTATION STATUS' versus Clinical Feature #10: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 68 | 2.3 (5.5) |
| SMAD2 MUTATED | 5 | 0.0 (0.0) |
| SMAD2 WILD-TYPE | 63 | 2.5 (5.7) |
Figure S1. Get High-res Image Gene #11: 'SMAD2 MUTATION STATUS' versus Clinical Feature #10: 'NUMBER.OF.LYMPH.NODES'
P value = 0.000874 (t-test), Q value = 0.14
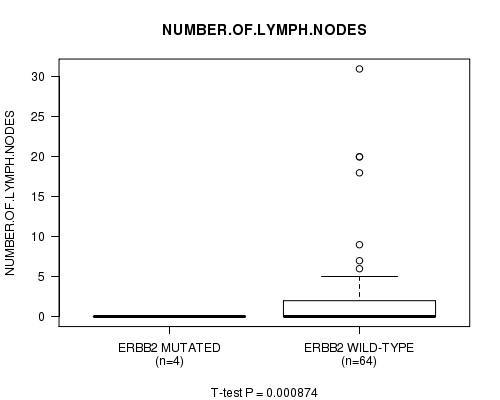
Table S2. Gene #13: 'ERBB2 MUTATION STATUS' versus Clinical Feature #10: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 68 | 2.3 (5.5) |
| ERBB2 MUTATED | 4 | 0.0 (0.0) |
| ERBB2 WILD-TYPE | 64 | 2.5 (5.7) |
Figure S2. Get High-res Image Gene #13: 'ERBB2 MUTATION STATUS' versus Clinical Feature #10: 'NUMBER.OF.LYMPH.NODES'
-
Mutation data file = READ-TP.mutsig.cluster.txt
-
Clinical data file = READ-TP.clin.merged.picked.txt
-
Number of patients = 69
-
Number of significantly mutated genes = 16
-
Number of selected clinical features = 10
-
Exclude genes that fewer than K tumors have mutations, K = 3
For survival clinical features, the Kaplan-Meier survival curves of tumors with and without gene mutations were plotted and the statistical significance P values were estimated by logrank test (Bland and Altman 2004) using the 'survdiff' function in R
For continuous numerical clinical features, two-tailed Student's t test with unequal variance (Lehmann and Romano 2005) was applied to compare the clinical values between tumors with and without gene mutations using 't.test' function in R
For binary or multi-class clinical features (nominal or ordinal), two-tailed Fisher's exact tests (Fisher 1922) were used to estimate the P values using the 'fisher.test' function in R
For multiple hypothesis correction, Q value is the False Discovery Rate (FDR) analogue of the P value (Benjamini and Hochberg 1995), defined as the minimum FDR at which the test may be called significant. We used the 'Benjamini and Hochberg' method of 'p.adjust' function in R to convert P values into Q values.
This is an experimental feature. The full results of the analysis summarized in this report can be downloaded from the TCGA Data Coordination Center.