This pipeline computes the correlation between significant arm-level copy number variations (cnvs) and selected clinical features.
Testing the association between copy number variation 25 arm-level results and 5 clinical features across 154 patients, 4 significant findings detected with Q value < 0.25.
-
1q gain cnv correlated to 'NUMBER.OF.LYMPH.NODES'.
-
3p gain cnv correlated to 'NUMBER.OF.LYMPH.NODES'.
-
5q loss cnv correlated to 'NUMBER.OF.LYMPH.NODES'.
-
20p loss cnv correlated to 'NUMBER.OF.LYMPH.NODES'.
Table 1. Get Full Table Overview of the association between significant copy number variation of 25 arm-level results and 5 clinical features. Shown in the table are P values (Q values). Thresholded by Q value < 0.25, 4 significant findings detected.
|
Clinical Features |
Time to Death |
AGE |
RADIATIONS RADIATION REGIMENINDICATION |
COMPLETENESS OF RESECTION |
NUMBER OF LYMPH NODES |
||
| nCNV (%) | nWild-Type | logrank test | t-test | Fisher's exact test | Fisher's exact test | t-test | |
| 1q gain | 0 (0%) | 150 |
1 (1.00) |
0.302 (1.00) |
1 (1.00) |
0.228 (1.00) |
0.00151 (0.187) |
| 3p gain | 0 (0%) | 150 |
1 (1.00) |
0.648 (1.00) |
1 (1.00) |
0.604 (1.00) |
0.00151 (0.187) |
| 5q loss | 0 (0%) | 151 |
1 (1.00) |
0.217 (1.00) |
1 (1.00) |
1 (1.00) |
0.00151 (0.187) |
| 20p loss | 0 (0%) | 150 |
1 (1.00) |
0.455 (1.00) |
0.125 (1.00) |
0.228 (1.00) |
0.00151 (0.187) |
| 3q gain | 0 (0%) | 149 |
1 (1.00) |
0.362 (1.00) |
1 (1.00) |
1 (1.00) |
0.439 (1.00) |
| 7p gain | 0 (0%) | 140 |
1 (1.00) |
0.0192 (1.00) |
1 (1.00) |
0.774 (1.00) |
0.17 (1.00) |
| 7q gain | 0 (0%) | 142 |
1 (1.00) |
0.0695 (1.00) |
1 (1.00) |
1 (1.00) |
0.958 (1.00) |
| 8p gain | 0 (0%) | 147 |
1 (1.00) |
0.636 (1.00) |
1 (1.00) |
0.405 (1.00) |
0.685 (1.00) |
| 8q gain | 0 (0%) | 140 |
1 (1.00) |
0.694 (1.00) |
1 (1.00) |
0.427 (1.00) |
0.856 (1.00) |
| 9p gain | 0 (0%) | 151 |
1 (1.00) |
0.725 (1.00) |
1 (1.00) |
0.555 (1.00) |
|
| 9q gain | 0 (0%) | 150 |
1 (1.00) |
0.7 (1.00) |
1 (1.00) |
1 (1.00) |
0.555 (1.00) |
| 10q gain | 0 (0%) | 151 |
1 (1.00) |
0.146 (1.00) |
1 (1.00) |
1 (1.00) |
0.456 (1.00) |
| 6q loss | 0 (0%) | 147 |
1 (1.00) |
0.261 (1.00) |
1 (1.00) |
1 (1.00) |
0.609 (1.00) |
| 8p loss | 0 (0%) | 115 |
1 (1.00) |
0.0895 (1.00) |
0.331 (1.00) |
0.495 (1.00) |
0.0935 (1.00) |
| 8q loss | 0 (0%) | 150 |
1 (1.00) |
0.651 (1.00) |
1 (1.00) |
1 (1.00) |
0.248 (1.00) |
| 10p loss | 0 (0%) | 150 |
1 (1.00) |
0.674 (1.00) |
1 (1.00) |
1 (1.00) |
0.875 (1.00) |
| 10q loss | 0 (0%) | 150 |
1 (1.00) |
0.395 (1.00) |
1 (1.00) |
0.604 (1.00) |
0.328 (1.00) |
| 12p loss | 0 (0%) | 147 |
1 (1.00) |
0.671 (1.00) |
1 (1.00) |
0.405 (1.00) |
0.142 (1.00) |
| 13q loss | 0 (0%) | 143 |
1 (1.00) |
0.819 (1.00) |
1 (1.00) |
1 (1.00) |
0.1 (1.00) |
| 16q loss | 0 (0%) | 134 |
1 (1.00) |
0.286 (1.00) |
1 (1.00) |
0.638 (1.00) |
0.207 (1.00) |
| 17p loss | 0 (0%) | 135 |
1 (1.00) |
0.464 (1.00) |
1 (1.00) |
0.517 (1.00) |
0.129 (1.00) |
| 18p loss | 0 (0%) | 140 |
1 (1.00) |
0.861 (1.00) |
1 (1.00) |
0.544 (1.00) |
0.487 (1.00) |
| 18q loss | 0 (0%) | 134 |
1 (1.00) |
0.72 (1.00) |
1 (1.00) |
0.517 (1.00) |
0.242 (1.00) |
| 21q loss | 0 (0%) | 151 |
1 (1.00) |
0.405 (1.00) |
1 (1.00) |
1 (1.00) |
0.503 (1.00) |
| 22q loss | 0 (0%) | 150 |
1 (1.00) |
0.467 (1.00) |
0.125 (1.00) |
0.228 (1.00) |
0.511 (1.00) |
P value = 0.00151 (t-test), Q value = 0.19
Table S1. Gene #1: '1q gain' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 135 | 0.2 (0.7) |
| 1Q GAIN CNV | 4 | 0.0 (0.0) |
| 1Q GAIN WILD-TYPE | 131 | 0.2 (0.8) |
Figure S1. Get High-res Image Gene #1: '1q gain' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
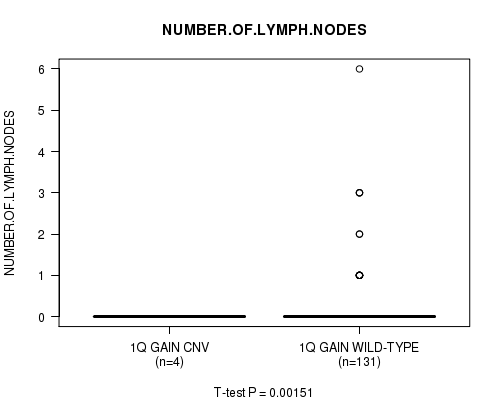
P value = 0.00151 (t-test), Q value = 0.19
Table S2. Gene #2: '3p gain' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 135 | 0.2 (0.7) |
| 3P GAIN CNV | 4 | 0.0 (0.0) |
| 3P GAIN WILD-TYPE | 131 | 0.2 (0.8) |
Figure S2. Get High-res Image Gene #2: '3p gain' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
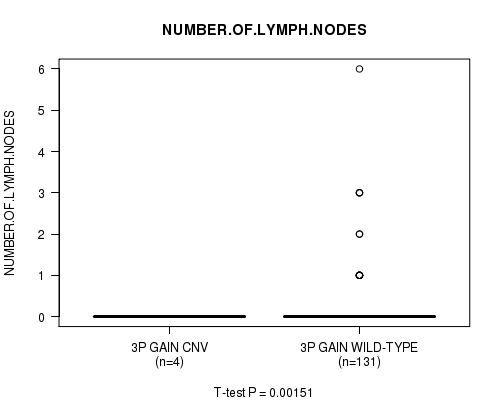
P value = 0.00151 (t-test), Q value = 0.19
Table S3. Gene #11: '5q loss' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 135 | 0.2 (0.7) |
| 5Q LOSS CNV | 3 | 0.0 (0.0) |
| 5Q LOSS WILD-TYPE | 132 | 0.2 (0.8) |
Figure S3. Get High-res Image Gene #11: '5q loss' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
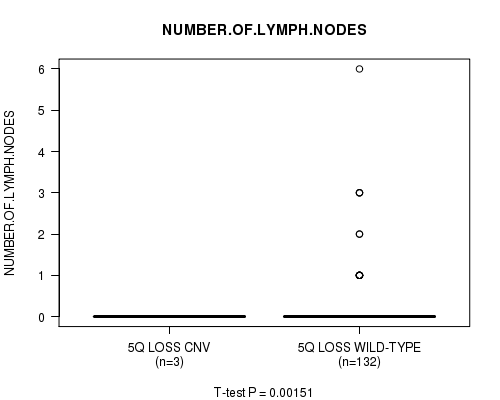
P value = 0.00151 (t-test), Q value = 0.19
Table S4. Gene #23: '20p loss' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
| nPatients | Mean (Std.Dev) | |
|---|---|---|
| ALL | 135 | 0.2 (0.7) |
| 20P LOSS CNV | 4 | 0.0 (0.0) |
| 20P LOSS WILD-TYPE | 131 | 0.2 (0.8) |
Figure S4. Get High-res Image Gene #23: '20p loss' versus Clinical Feature #5: 'NUMBER.OF.LYMPH.NODES'
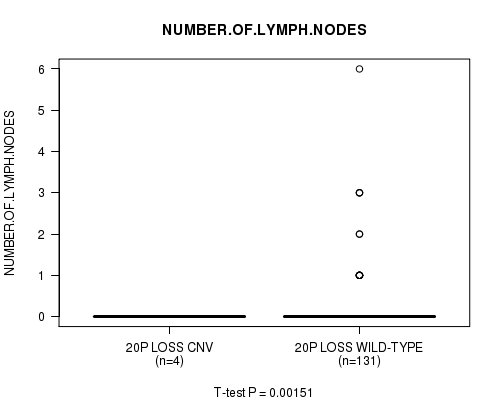
-
Mutation data file = broad_values_by_arm.mutsig.cluster.txt
-
Clinical data file = PRAD-TP.clin.merged.picked.txt
-
Number of patients = 154
-
Number of significantly arm-level cnvs = 25
-
Number of selected clinical features = 5
-
Exclude genes that fewer than K tumors have mutations, K = 3
For survival clinical features, the Kaplan-Meier survival curves of tumors with and without gene mutations were plotted and the statistical significance P values were estimated by logrank test (Bland and Altman 2004) using the 'survdiff' function in R
For continuous numerical clinical features, two-tailed Student's t test with unequal variance (Lehmann and Romano 2005) was applied to compare the clinical values between tumors with and without gene mutations using 't.test' function in R
For binary or multi-class clinical features (nominal or ordinal), two-tailed Fisher's exact tests (Fisher 1922) were used to estimate the P values using the 'fisher.test' function in R
For multiple hypothesis correction, Q value is the False Discovery Rate (FDR) analogue of the P value (Benjamini and Hochberg 1995), defined as the minimum FDR at which the test may be called significant. We used the 'Benjamini and Hochberg' method of 'p.adjust' function in R to convert P values into Q values.
This is an experimental feature. The full results of the analysis summarized in this report can be downloaded from the TCGA Data Coordination Center.