This pipeline computes the correlation between significant copy number variation (cnv focal) genes and molecular subtypes.
Testing the association between copy number variation 2 focal events and 8 molecular subtypes across 66 patients, 2 significant findings detected with P value < 0.05 and Q value < 0.25.
-
amp_8q11.23 cnv correlated to 'MRNASEQ_CNMF'.
-
amp_15q22.31 cnv correlated to 'MRNASEQ_CNMF'.
Table 1. Get Full Table Overview of the association between significant copy number variation of 2 focal events and 8 molecular subtypes. Shown in the table are P values (Q values). Thresholded by P value < 0.05 and Q value < 0.25, 2 significant findings detected.
|
Clinical Features |
CN CNMF |
METHLYATION CNMF |
MRNASEQ CNMF |
MRNASEQ CHIERARCHICAL |
MIRSEQ CNMF |
MIRSEQ CHIERARCHICAL |
MIRSEQ MATURE CNMF |
MIRSEQ MATURE CHIERARCHICAL |
||
| nCNV (%) | nWild-Type | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | Fisher's exact test | |
| amp 8q11 23 | 19 (29%) | 47 |
0.321 (1.00) |
0.625 (1.00) |
0.00283 (0.0453) |
0.0245 (0.343) |
1 (1.00) |
0.228 (1.00) |
0.363 (1.00) |
0.118 (1.00) |
| amp 15q22 31 | 23 (35%) | 43 |
0.0534 (0.694) |
0.582 (1.00) |
0.00627 (0.094) |
0.182 (1.00) |
0.526 (1.00) |
0.301 (1.00) |
0.554 (1.00) |
0.365 (1.00) |
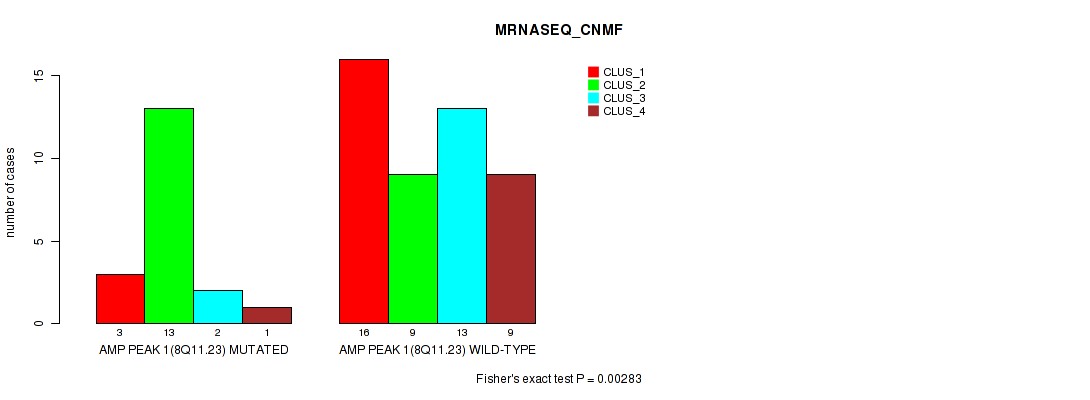
P value = 0.00283 (Fisher's exact test), Q value = 0.045
Table S1. Gene #1: 'amp_8q11.23' versus Molecular Subtype #3: 'MRNASEQ_CNMF'
| nPatients | CLUS_1 | CLUS_2 | CLUS_3 | CLUS_4 |
|---|---|---|---|---|
| ALL | 19 | 22 | 15 | 10 |
| AMP PEAK 1(8Q11.23) MUTATED | 3 | 13 | 2 | 1 |
| AMP PEAK 1(8Q11.23) WILD-TYPE | 16 | 9 | 13 | 9 |
Figure S1. Get High-res Image Gene #1: 'amp_8q11.23' versus Molecular Subtype #3: 'MRNASEQ_CNMF'
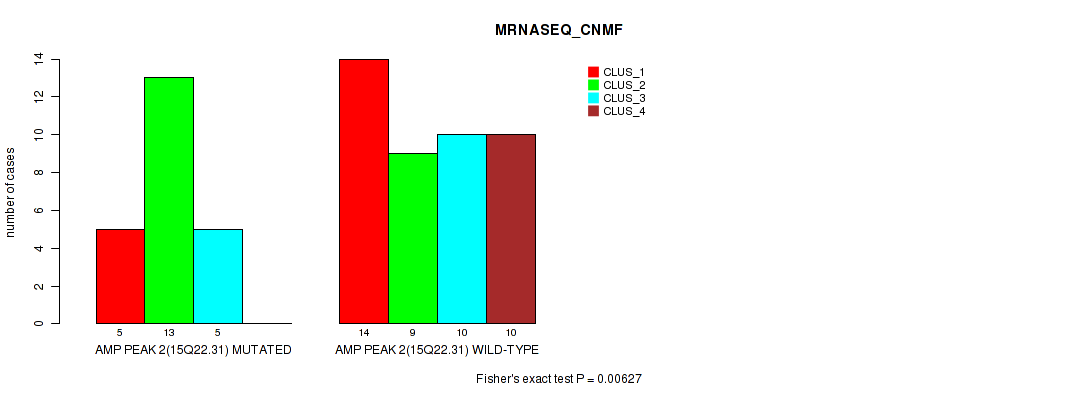
P value = 0.00627 (Fisher's exact test), Q value = 0.094
Table S2. Gene #2: 'amp_15q22.31' versus Molecular Subtype #3: 'MRNASEQ_CNMF'
| nPatients | CLUS_1 | CLUS_2 | CLUS_3 | CLUS_4 |
|---|---|---|---|---|
| ALL | 19 | 22 | 15 | 10 |
| AMP PEAK 2(15Q22.31) MUTATED | 5 | 13 | 5 | 0 |
| AMP PEAK 2(15Q22.31) WILD-TYPE | 14 | 9 | 10 | 10 |
Figure S2. Get High-res Image Gene #2: 'amp_15q22.31' versus Molecular Subtype #3: 'MRNASEQ_CNMF'
-
Copy number data file = transformed.cor.cli.txt
-
Molecular subtype file = KICH-TP.transferedmergedcluster.txt
-
Number of patients = 66
-
Number of significantly focal cnvs = 2
-
Number of molecular subtypes = 8
-
Exclude genes that fewer than K tumors have alterations, K = 3
For binary or multi-class clinical features (nominal or ordinal), two-tailed Fisher's exact tests (Fisher 1922) were used to estimate the P values using the 'fisher.test' function in R
For multiple hypothesis correction, Q value is the False Discovery Rate (FDR) analogue of the P value (Benjamini and Hochberg 1995), defined as the minimum FDR at which the test may be called significant. We used the 'Benjamini and Hochberg' method of 'p.adjust' function in R to convert P values into Q values.
In addition to the links below, the full results of the analysis summarized in this report can also be downloaded programmatically using firehose_get, or interactively from either the Broad GDAC website or TCGA Data Coordination Center Portal.