A TCGA sample is profiled to detect the copy number variations and expressions of genes. This pipeline attempts to correlate copy number and expression data of genes across samples to determine if the copy number variations also result in differential expressions. This report contains the calculated correlation coefficients based on measurements of genomic copy number (log2) values and intensity of the expressions of the corresponding feature across patients. High positive/low negative correlation coefficients indicate that genomic alterations result in differences in the expressions of mRNA the genomic regions transcribe.
The correlation coefficients in 10, 20, 30, 40, 50, 60, 70, 80, 90 percentiles are -0.0124, 0.02434, 0.06, 0.0979, 0.13795, 0.17972, 0.2217, 0.2684, 0.33793, respectively.
Number of genes and samples used for the calculation are shown in Table 1. Figure 1 shows the distribution of calculated correlation coefficients and quantile-quantile plot of the calculated correlation coefficients against a normal distribution. Table 2 shows the top 20 features ordered by the value of correlation coefficients.
Table 1. Counts of mRNA and number of samples in copy number and expression data sets and common to both
| Category | Copy number | Expression | Common |
|---|---|---|---|
| Sample | 1090 | 528 | 492 |
| Genes | 24771 | 12043 | 10838 |
Figure 1. Summary figures. Left: histogram showing the distribution of the calculated correlations across samples for all Genes. Right: QQ plot of the calculated correlations across samples. The QQ plot is used to plot the quantiles of the calculated correlation coefficients against that derived from a normal distribution. Points deviating from the blue line indicate deviation from normality.
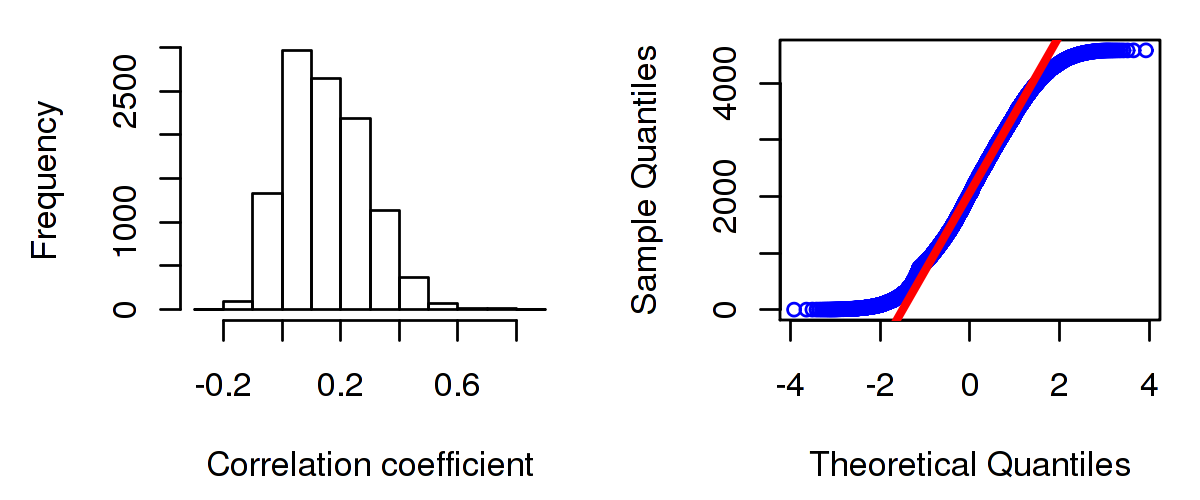
Table 2. Get Full Table Top 20 features (defined by the feature column) ranked by correlation coefficients
| feature | r | p-value | q-value | chrom | start | end | geneid |
|---|---|---|---|---|---|---|---|
| SEC61G | 0.836 | 0 | 0 | 23480 | 7p11.2 | 1.267 | 3.657 |
| METTL1 | 0.8304 | 0 | 0 | 4234 | 12q14.1 | 0.335 | 3.657 |
| CDK4 | 0.8064 | 0 | 0 | 1019 | 12q14.1 | 0.335 | 3.657 |
| TSPAN31 | 0.796 | 0 | 0 | 6302 | 12q14.1 | 0.335 | 3.657 |
| TSFM | 0.795 | 0 | 0 | 10102 | 12q14.1 | 0.335 | 3.657 |
| MDM2 | 0.7849 | 0 | 0 | 4193 | 12q15 | 0.335 | 0.000 |
| MRPS17 | 0.7604 | 0 | 0 | -2505 | 7p11.2 | 1.267 | 0.813 |
| EGFR | 0.7567 | 0 | 0 | 1956 | 7p11.2 | 1.267 | 3.657 |
| CDKN2A | 0.7545 | 0 | 0 | 1029 | 9p21.3 | -1.106 | 0.001 |
| LANCL2 | 0.7323 | 0 | 0 | 55915 | 7p11.2 | 1.267 | 0.813 |
| CYP27B1 | 0.7167 | 0 | 0 | 1594 | 12q14.1 | 0.335 | 3.657 |
| CCT6A | 0.7156 | 0 | 0 | 908 | 7p11.2 | 1.267 | 0.813 |
| DCTN2 | 0.7107 | 0 | 0 | 10540 | 12q13.3 | 0.335 | 3.657 |
| CHIC2 | 0.7009 | 0 | 0 | 26511 | 4q12 | -0.077 | -0.008 |
| CTDSP2 | 0.6882 | 0 | 0 | 10106 | 12q14.1 | 0.335 | 3.657 |
| ETNK2 | 0.6878 | 0 | 0 | 55224 | 1q32.1 | 0.232 | 0.008 |
| CHCHD2 | 0.6833 | 0 | 0 | 51142 | 7p11.2 | 1.267 | 0.813 |
| KLHL9 | 0.6696 | 0 | 0 | -3036 | 9p21.3 | -0.044 | 0.001 |
| MDM4 | 0.6694 | 0 | 0 | 4194 | 1q32.1 | 0.232 | 0.008 |
| GBAS | 0.6651 | 0 | 0 | 2631 | 7p11.2 | 1.267 | 0.813 |
Gene level (TCGA Level III) expression data and copy number data of the corresponding loci derived by using the CNTools package of Bioconductor were used for the calculations. Pearson correlation coefficients were calculated for each pair of genes shared by the two data sets across all the samples that were common. The input file "*.medianexp.txt" is generated in the pipeline mRNA_Preprocess_Median in the stddata run.
Pairwise correlations between the log2 copy numbers and expressions of each gene across samples were calculated using Pearson correlation.
In addition to the links below, the full results of the analysis summarized in this report can also be downloaded programmatically using firehose_get, or interactively from either the Broad GDAC website or TCGA Data Coordination Center Portal.