APOBEC (Apolipoprotein B Editing Complex Like Polypeptide) cytidine deaminases normally serve to restrict retroelements (retroviruses and retrotransposons) by removing an amino group from cytidines in single stranded (ss) DNA formed during the retroelement's replication cycle. By accessing ssDNAs formed during replication, repair or transcription in chromosomes, APOBECs can cause mutations in human cancer genomes. So far, the APOBEC3B and APOBEC3A proteins are the main candidates for inducing mutation in human cancers, however other APOBECs from this subclass or even other APOBEC-like proteins also could be involved. The mutagenesis signature of these enzymes has been recently found in multiple types of human cancers [1][2][3]. The fraction of APOBEC signature mutations in a cancer exome can be as high as 70% of the entire mutation load and the number of such mutations can be over 1,000. These findings have led to the general question about presence of APOBEC mutagenesis pattern in various sequenced cancer genomes or exomes. Because of APOBECs' sequence- and ssDNA substrate-specificity, statistical analysis can be performed for a single defined mechanism-based hypothesis. This provides sufficient statistical power to highlight and evaluate the pattern within a complex mix of mutagenic processes operating throughout the history of individual cancers. Moreover, statistical evaluation can be done for each individual sample allowing the identification of individual tumors that contain significant presence of the APOBEC mutagenesis pattern, which in turn enables downstream exploration for correlations between this mutagenesis and tumor genotype and phenotype.
Components of the Pattern of Mutagenesis by APOBEC Cytidine Deaminases (P-MACD) analysis:
-
The genome- or exome-wide prevalence of the APOBEC mutagenesis signature and the enrichment of this signature over its presence expected for random mutagenesis. APOBECs deaminate cytidines predominantly in a tCw motif. The APOBEC mutagenesis signature is composed of approximately equal numbers of two kinds of changes in this motif – tCw→tTw and tCw→tGw mutations (flanking nucleotides shown in small letters; w=A or T). tCw→tAw changes are not included into P-MACD analysis, because cytosine deamination in ssDNA predominantly results in changes to thymine or guanine as compared to changes to adenine. P-MACD calculates on a per sample basis, the enrichment of the APOBEC mutation signature among all mutated cytosines in comparison to the fraction of cytosines that occur in the tCw motif among the +/- 20 nucleotides surrounding each mutated cytosine. In addition, P-MACD calculates other parameters that characterize the prevalence of the APOBEC mutagenesis pattern in a sample and/or that are useful for downstream analyses and comparisons. The results of the APOBEC mutagenesis signature analyses are presented on Figures 1–5.
-
The presence of C- or G-coordinated clusters of closely spaced mutations. C- or G-coordinated mutation clusters contain stretches of mutations, each separated by <10,000 nt, that either occur only at cytosines or only in guanines of the same strand within a given cluster. Moreover, the probability of producing the spatial distribution of mutations in these clusters by random distribution of all the mutations estimated to occur within the genome is < 1E-4. C- or G-coordinated clusters are enriched with APOBEC signature mutations. The presence of such clusters supports a mechanism of mutagenesis via cytidine-specific lesions in transient stretches of ssDNA. Cluster analysis works the best for whole-genome sequence (WGS) data, but can have sufficient statistical power to be utilized with whole-exome sequencing (WES). The results of cluster analysis are shown on Figures 6–9.
Both parts of analysis are described in detail in reference [3]
There are 978 tumor samples in this analysis. The Benjamini-Hochberg-corrected p-value for enrichment of the APOBEC mutation signature in 227 samples is <=0.05. Out of these, 220 have enrichment values >2, which implies that in such samples at least 50% of APOBEC signature mutations have been in fact made by APOBEC enzyme(s).
Column content and calculation of values referred in the legends to all figures below are described in “Readme_columns_in_sum_files.txt” file (see hyperlink in the Output section.)
Figure 1. Get High-res Image Fold-enrichment of APOBEC mutagenesis signature over the expected occurrence for random mutagenesis. Fold enrichment values are taken from the “APOBEC_enrich” column and Benjamini-Hochberg-corrected p-values are taken from the “BH_Fisher_p-value_tCw” column in the “*_sorted_sum_all_fisher_Pcorr.txt” file. Samples are categorized into color coded bins based on their q-values and fold enrichment values. Fold enrichment bin sizes are increments of 1 unit. All samples displaying a q-value >0.05 are placed in one bin (black) regardless of the fold APOBEC enrichment in the sample. The maximum fold enrichment for each bin is indicated in the figure legend, with the number of samples in its category shown in parentheses.

Figure 2. Get High-res Image Fold enrichment of the APOBEC mutagenesis signature over the expected occurrence for random mutagenesis in individual samples. Fold enrichment values are taken from the “APOBEC_enrich” column and Benjamini-Hochberg-corrected p-values are taken from the “BH_Fisher_p-value_tCw” column in “*_sorted_sum_all_fisher_Pcorr.txt” file. See also legend to Figure 1.
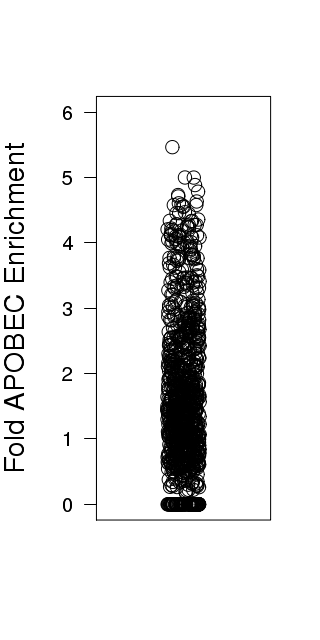
Figure 3. Get High-res Image Relative load of APOBEC signature mutations. The Fraction of Total Mutations values are taken from the “[tCw_to_G+tCw_to_T]_per_mut” column in the “*_sorted_sum_all_fisher_Pcorr.txt” file.
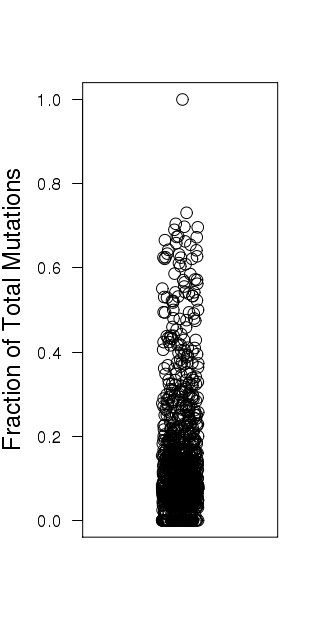
Figure 4. Get High-res Image Numbers of APOBEC and non-APOBEC mutations (X-axis) in each sample (Y-axis). All values are calculated based on the values in the columns “indels”, “substitutions”, and columns containing individual substitution types in the “*_sorted_sum_all_fisher_Pcorr.txt” file. “APOBEC to G” – number of tCw→tGw mutations; “APOBEC to T” – number of tCw→tTw mutations; “non-APOBEC C:G” – number of mutations in C:G base pairs not conforming to the APOBEC signature (= total of tCw→tAw plus all mutations in C not occurring in the tCw motif); “A:T” –- all mutations in A:T base pairs; “indels” – all indels. All counts include complementary mutations. Samples are ordered by total mutation counts in the descending order. Since only base substitutions are shown, bars with excessive numbers of non-substitution mutations may be shorter than the two flanking bars.

Figure 5. Get High-res Image Minimum estimate of the number of APOBEC induced mutations in a sample. “APOBEC_MutLoad_MinEstimate” is calculated using the formula: [“tCw_to_G+tCw_to_T”]*[(“APOBEC_enrich”-1)/“APOBEC_enrich”] to determine the number of APOBEC signature mutations in excess of what would be expected by random mutagenesis. Calculated values are rounded to the nearest whole number. “APOBEC_MutLoad_MinEstimate” is calculated only for samples with a “BH_Fisher_p-value_tCw” value less than or equal to 0.05, signifying a statistical over-representation of APOBEC mutagenesis. Samples with “BH_Fisher_p-value_tCw” value greater than 0.05 receive a value of 0. “APOBEC_MutLoad_MinEstimate” is plotted on a logarithmic scale (with a pseudocount of 1) for better visualization of values in all sections of its range.

Figure 6. Get High-res Image Numbers of different types of mutation clusters. Values are taken from columns with corresponding names in the file “*_sorted_sum_clusters.txt”.

Figure 7. Get High-res Image The number of mutations within C- or G-coordinated clusters occurring in different known C- or G-specific mutation motifs. Counts are totaled among all samples. “C/G” – any mutation in C:G base pairs; “TC/GA” – any mutation in a less stringent APOBEC motif: tC (mutated nucleotide capitalized); “TCW/WGA” – any mutation in the stringent APOBEC motif: tCw; “WRC/GYW” – any mutation in the wrC motif for AID cytidine deaminase (r=A or G; y=C or T); “CC/GG” – any mutation in the cC motif for APOBEC3G; “CG/CG” – any mutation in the Cg (or CpG) motif frequently methylated (5me-C) and prone to deamination. Fold enrichments are calculated using values from the corresponding columns (including complementary mutations) in the “Totals” row of the “*_sorted_sum_G_C_clusters.txt” file. Fold enrichments for each signature (shown as numbers above the bars) are calculated the same as the fold enrichment for the APOBEC mutation motif. All three possible substitutions of C are included (i.e. to A, T and G). When tCw-specific APOBECs are the source of clustered mutations, both tC and tCw should be enriched with the latter being greater. Moreover, other C-specific mutation signatures should be depleted.

Figure 8. Get High-res Image The number of mutations categorized into three possible types of substitutions at the tCw motif (complementary mutations included) within C- or G-coordinated clusters. Numbers are calculated using the “Totals” row in the corresponding columns of the “*_sorted_sum_G_C_clusters.txt” file. Mutations caused by cytidine deamination in stretches of ssDNA caused by APOBEC mutagenesis are expected to be predominantly C-->T or C-->G with very few C-->A. Base substitution categories are shown within TCW context.

Figure 9.
Get High-res Image
Fold enrichment of the APOBEC mutation signature in clusters of different sizes of two categories – C- or G-coordinated or non-coordinated as well as in non-clustered (scattered) mutations. Smaller clusters usually show less enrichment because they have a higher chance to be formed by random mutations that occurred in close vicinity to each other. Values above the bars display the total number of mutation clusters in each class and in parentheses the total numbers of APOBEC signature mutations in a category. Values for fold enrichment as well as mutation and mutation cluster counts are taken from the following output files which can be downloaded from firehose_get (https://confluence.broadinstitute.org/display/GDAC/Download):
“*_sorted_sum10c.txt” for scattered mutations;
“*_sorted_sum04a.txt” for C- or G-coordinated clusters with 2 mutations;
“*_sorted_sum04b.txt” for C- or G-coordinated clusters with 3 mutations;
“*_sorted_sum04f.txt” for C- or G-coordinated clusters with 4 mutations;
“*_sorted_sum04h.txt” for C- or G-coordinated clusters with 5 mutations;
“*_sorted_sum04i.txt” for C- or G-coordinated clusters with >5 mutations;
“*_sorted_sum01a.txt” for non-coordinated clusters with 2 mutations;
“*_sorted_sum01b.txt” for non-coordinated clusters with 3 mutations;
“*_sorted_sum01f.txt” for non-coordinated clusters with 4 mutations;
“*_sorted_sum01h.txt” for non-coordinated clusters with 5 mutations;
“*_sorted_sum01i.txt” for non-coordinated clusters with >5 mutations;
and are compiled in the file with the summary of calculation results “*_sorted_sum_APOBECenrich_ClustSize.txt”.

-
Mutation Annotation File (MAF): tab-delimited table of tumor-specific mutation calls per row in The Cancer Genome Atlas (TCGA) format, based on either whole exome sequenciong (WES) or whole genome sequencing (WGS). Non-ASCII binary characters (>127 decimal) are not allowed in any column. The file is not required to be a TCGA MAF, however the following column names and the value syntax in these columns should be fixed:
-
Sample ID – column name: “Tumor_Sample_Barcode”, value syntax not defined, each sample ID must be unique;
-
Chromosome – column name: “Chromosome”; value syntax: 1-22, X, Y.
(Note that P-MACD filters out mutation calls for which chromosome name is not one of the 24 standard human chromosomes, such as mitochondrial DNA or unassigned chromosomal fragments.) -
Position – column name: “Start_position”; value syntax: integer numbers
-
Reference allele – column name: “Reference_Allele”; value syntax; A, T, C, G (nucleotides in capitals)
-
Tumor allele – column name: “Tumor_Seq_Allele2”; value syntax: A, T, C, G (nucleotides in capitals)
-
Variant type – column name: “Variant_Type”; value syntax: SNP (single base substitutions), other – not defined
-
Reference Genome: nucleotide sequence of the reference genome with one entry per chromosome in FASTA format.
-
Fraction of Genome Sequenced: 0.01 for whole-exome MAF and 1.0 for whole-genome MAF. (Used only to identify mutation clusters).
This run of P-MACD was executed with the following parameter values as inputs:
-
Mutation Annotation File = Mutsig_maf_modified.maf.txt
-
Reference Genome = /xchip/cga/reference/hg19/hg19canonical.fa
-
Fraction of Genome Sequenced = 0.01
A detailed description of the output files is provided in the Readme files:
-
“Readme_content_of_files.txt” file: provides a description of the content of each file in the output, identified by a content specific suffix following the name of the original MAF file.
-
“Readme_columns_in_anz_MAF_files.txt” file: describes the columns that P-MACD adds to the input MAF file.
-
“Readme_columns_in_sum_files.txt” file: describes the columns in all “*_sorted_sum*.txt” files.
-
“*_List_of_inputs.txt” file: provides the information about the MAF file and input values specific for a run and provided by the user;
There are two main parts of the output:
MAF files “*_sorted_anz*.txt”
which contain all columns of the initial MAF file with rows sorted by “Tumor_Sample_Barcode”, then by “Chromosome”, then by “Start_position”. P-MACD removes duplicate mutations if multiple rows exist that contain identical values within the “Tumor_Sample_Barcode”, “Chromosome”, “Start_position”, “Reference_Allele”, and “Variant_Type” columns. Duplicates occur when a single MAF contains mutations identified by comparing the sequence of one tumor sample to both whole blood and adjacent normal tissue controls. P-MACD retains the first occurrence of the duplicated mutation in the sorted MAF. The output MAFs contain also several additional columns annotating each mutation based on whether it fits the APOBEC mutation signature (columns: APOBEC_mutation, APOBEC_mutation_to_G, and APOBEC_mutation_to_T) and if it occurs in a cluster of tightly spaced mutations. Groups of very tightly spaced mutations with inter-mutation distances <10 nt, which are likely to arise from a mutagenesis event triggered by trans-lesion synthesis across a single DNA lesion, are also highlighted. Such events are called complex and counted as a single mutation when calculating cluster p-values. Annotating mutations by the APOBEC signature highlights mutations that were likely caused by APOBEC mutagenesis. Such separation should be done only in samples with a statistically significant fold enrichment and large effect size. For example, if a sample has a statistically significant 2-fold enrichment with the APOBEC mutagenesis pattern, then at least 50% of APOBEC signature mutations could be expected to be really caused by APOBEC cytidine deamination.
Summary files “*_sorted_sum*.txt”
There are two kinds of summary files:
A. Sample-specific summaries.
Each row in a summary file provides values calculated for a given sample. The last row provides totals among all samples. The summary files directly used for generating Figures 1–9 are listed in the legend(s) of the corresponding Figure(s). The following values (all present in “*_sorted_sum_all_fisher_Pcorr.txt” file) are the most useful for analyses investigating correlations between APOBEC mutagenesis and other features of cancer samples:
-
Fold-enrichment with APOBEC mutagenesis signature – “APOBEC_enrich” column;
-
Absolute number of APOBEC signature mutations in a sample – “tCw_to_G+tCw_to_T” column;
-
Fraction of APOBEC signature mutations in a sample – “[tCw_to_G+tCw_to_T]_per_mut” column;
-
Benjamini-Hochberg-corrected p-values – “BH_Fisher_p-value_tCw” column.
-
Minimum estimate of the number of APOBEC induced mutations in a sample – “APOBEC_MutLoad_MinEstimate” column
B. Summaries across all samples.
The file “*_sorted_sum_APOBECenrich_ClustSize.txt” contains results of calculations graphically represented in Figure 9.
In addition to the links below, the full results of the analysis summarized in this report can also be downloaded programmatically using firehose_get, or interactively from either the Broad GDAC website or TCGA Data Coordination Center Portal.