This pipeline inspects significant overlapping pathway gene sets for a given gene list using a hypergeometric test. For the gene set database, we uses GSEA MSigDB Class2: Canonical Pathways DB as a gene set data. Further details about the MsigDB gene sets, please visit The Broad Institute GSEA MsigDB
For a given gene list, a hypergeometric test was tried to find significant overlapping canonical pathways using 1320 gene sets. In terms of FDR adjusted p.values, top 5 significant overlapping gene sets are listed as below.
-
KEGG_GLIOMA, KEGG_MELANOMA, KEGG_PATHWAYS_IN_CANCER, KEGG_PROSTATE_CANCER, KEGG_NON_SMALL_CELL_LUNG_CANCER
Table 1. Get Full Table This table shows significant gene sets in which at least one gene is found and its FDR adjusted p.value is smaller than 0.3. the hypergeometric p-value is a probability of randomly drawing x or more successes(gene overlaps in gene set database) from the population (gene universe consisting of N number of genes) in k total draws(the number of input genes). The hypergeometric test is identical to the corresponding one-tailed version of Fisher's exact test. That is, P(X=x) = f(x| N,m,k). The FDR q.value was obtained for 1320 multiple comparison.
| GS(gene set) pathway name | gene.list | GS size (m) | n.NotInGS (n) | Gene universe (N) | n.drawn (k) | n.found (x) | p.value (p(X>=x)) | FDR (q.value) |
|---|---|---|---|---|---|---|---|---|
| KEGG GLIOMA | gene.list | 65 | 45891 | 45956 | 110 | 9 | 5.277e-14 | 6.966e-11 |
| KEGG MELANOMA | gene.list | 71 | 45885 | 45956 | 110 | 8 | 7.805e-12 | 5.152e-09 |
| KEGG PATHWAYS IN CANCER | gene.list | 328 | 45628 | 45956 | 110 | 12 | 2.691e-11 | 1.184e-08 |
| KEGG PROSTATE CANCER | gene.list | 89 | 45867 | 45956 | 110 | 8 | 5.000e-11 | 1.580e-08 |
| KEGG NON SMALL CELL LUNG CANCER | gene.list | 54 | 45902 | 45956 | 110 | 7 | 5.986e-11 | 1.580e-08 |
| PID ERBB1 RECEPTOR PROXIMAL PATHWAY | gene.list | 35 | 45921 | 45956 | 110 | 6 | 2.513e-10 | 5.528e-08 |
| REACTOME SIGNALING BY CONSTITUTIVELY ACTIVE EGFR | gene.list | 18 | 45938 | 45956 | 110 | 5 | 5.990e-10 | 1.130e-07 |
| KEGG SMALL CELL LUNG CANCER | gene.list | 84 | 45872 | 45956 | 110 | 7 | 1.443e-09 | 2.382e-07 |
| KEGG ENDOMETRIAL CANCER | gene.list | 52 | 45904 | 45956 | 110 | 6 | 3.049e-09 | 4.472e-07 |
| KEGG NEUROTROPHIN SIGNALING PATHWAY | gene.list | 126 | 45830 | 45956 | 110 | 7 | 2.478e-08 | 2.973e-06 |
| KEGG CHRONIC MYELOID LEUKEMIA | gene.list | 73 | 45883 | 45956 | 110 | 6 | 2.448e-08 | 2.973e-06 |
| PID PDGFRBPATHWAY | gene.list | 129 | 45827 | 45956 | 110 | 7 | 2.916e-08 | 3.208e-06 |
| BIOCARTA ARF PATHWAY | gene.list | 17 | 45939 | 45956 | 110 | 4 | 7.219e-08 | 6.813e-06 |
| REACTOME SIGNALING BY FGFR MUTANTS | gene.list | 44 | 45912 | 45956 | 110 | 5 | 7.226e-08 | 6.813e-06 |
| PID PI3KCIPATHWAY | gene.list | 49 | 45907 | 45956 | 110 | 5 | 1.257e-07 | 9.758e-06 |
| REACTOME SIGNALING BY FGFR1 FUSION MUTANTS | gene.list | 19 | 45937 | 45956 | 110 | 4 | 1.171e-07 | 9.758e-06 |
| REACTOME DOWNSTREAM SIGNAL TRANSDUCTION | gene.list | 95 | 45861 | 45956 | 110 | 6 | 1.198e-07 | 9.758e-06 |
| PID ECADHERIN KERATINOCYTE PATHWAY | gene.list | 21 | 45935 | 45956 | 110 | 4 | 1.802e-07 | 1.252e-05 |
| REACTOME SIGNALING BY ERBB2 | gene.list | 101 | 45855 | 45956 | 110 | 6 | 1.726e-07 | 1.252e-05 |
| PID PDGFRAPATHWAY | gene.list | 22 | 45934 | 45956 | 110 | 4 | 2.198e-07 | 1.451e-05 |
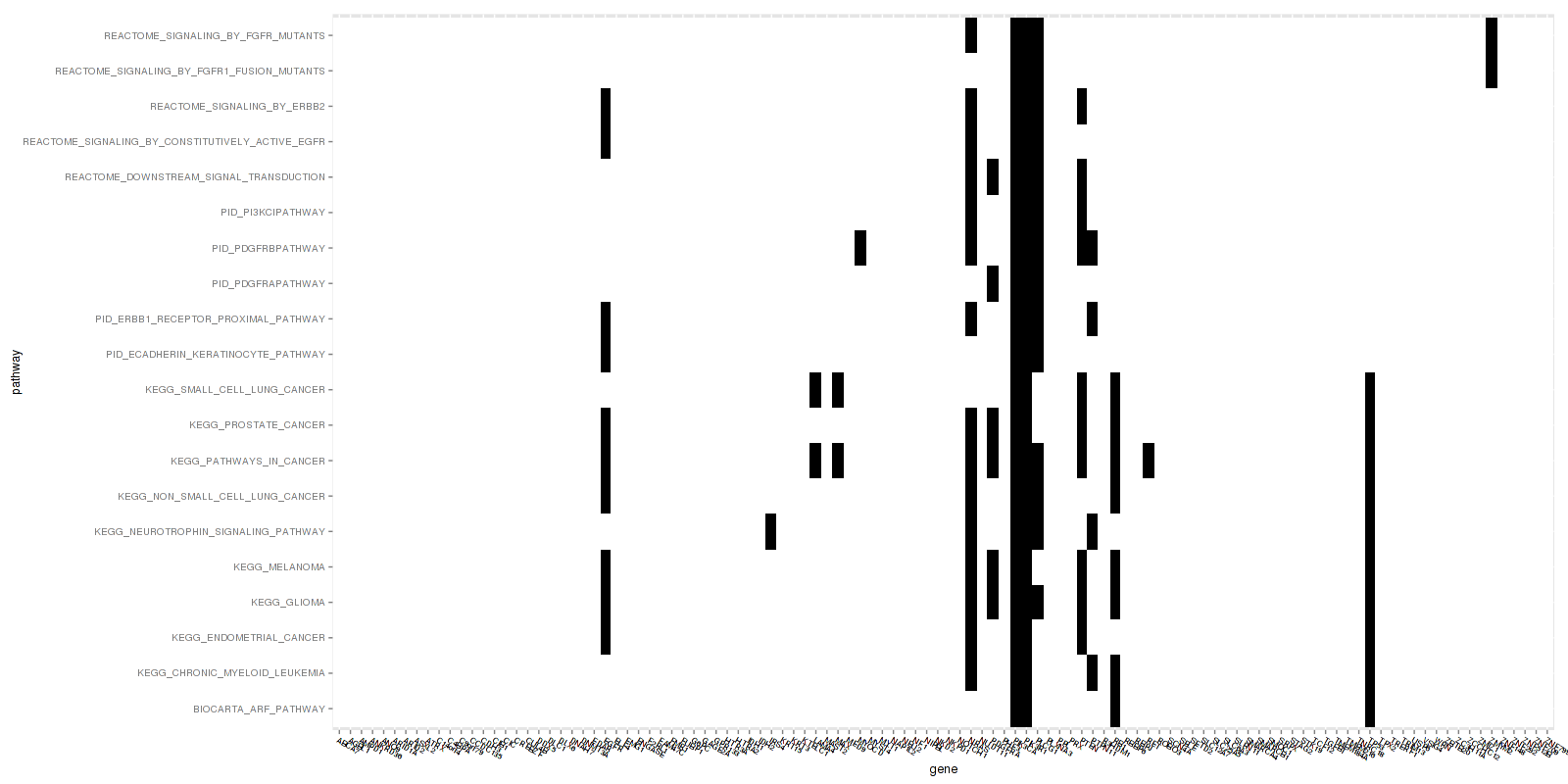
Figure 1. Get High-res Image This figure is an event heatmap indicating gene matches across gene sets
-
Gene set database = c2.cp.v4.0.symbols.gmt
-
Input gene list = MutSig2CV.input.genenames.txt
For a given gene list, it uses a hypergeometric test to get a significance of each overlapping pathway gene set. The hypergeometric p-value is obtained by R library function phyper() and is defined as a probability of randomly drawing x or more successes(gene matches) from the population consisting N genes in k(the input genes) total draws.
-
a cumulative p-value using the R function phyper():
-
ex). a probability to see at least x genes in the group is defined as p(X>=x) = 1 - p(X<=x)= 1 - phyper(x-1, m, n, k, lower.tail=FALSE, log.p=FALSE) that is, f(x| N, m, k) = (m) C (k) * ((N-m) C (n-k)) / ((N) C (n))
-
The hypergeometric test is identical to the corresponding one-tailed version of Fisher's exact test.
-
ex). Fisher' exact test = matrix(c(n.Found, n.GS-n.Found, n.drawn-n.Found, n.NotGS- (n.drawn-n.Found)), nrow=2, dimnames = list(inputGenes = c("Found", "NotFound"),GeneUniverse = c("GS", "nonGS")) )
In addition to the links below, the full results of the analysis summarized in this report can also be downloaded programmatically using firehose_get, or interactively from either the Broad GDAC website or TCGA Data Coordination Center Portal.